2022年5月4日,Nature在线发表了德国马克斯-普朗克煤炭研究所Benjamin List教授团队与香港中文大学(深圳)成贵娟教授团队题为“Organocatalytic stereoselective cyanosilylation of small ketones”的研究成果。
课题组通过设计并使用了一种仿酶穴状空腔结构的有机超强酸IDPi催化剂,成功实现了2-丁酮的不对称硅氰化反应,对映体过量值高达96%。理论计算揭示了反应机理和手性识别机制。论文通讯作者是Benjamin List教授和成贵娟教授;第一作者是周慧博士。
根据“锁-钥”催化模型,酶催化可以高效、立体专一性地实现化学转化,因而有着其他催化剂不可替代的、得天独厚的优势。二烷基酮由于两个取代基电性和空间位阻比较相似,难以被手性催化剂识别,因此其精准不对称诱导反应一直难以被解决。其中,2-丁酮参与的不对称反应由于甲基和乙基基团过于相似,往往只有酶催化剂有能力对其进行区分。受周其林院士课题组金属铱络合物仿酶催化2-丁酮的不对称氢化工作的启发(Nat. Catal.2020, 3, 621),近期,德国马克斯-普朗克煤炭研究所Benjamin List教授课题组设计并使用了一种仿酶穴状空腔结构的有机超强酸IDPi催化剂,成功实现了2-丁酮的不对称硅氰化反应,对映体过量值高达96%。
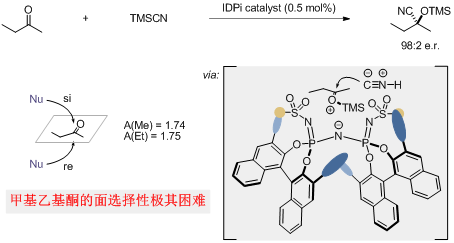
图1:研究背景和反应设计(图片来源:Nature)
在过去的近百年里,不对称硅氰化反应广为研究,已经比较成熟。然而由于2-丁酮中甲基和乙基较为相似,其在不对称反应方面的研究很难。目前仅有的几个成功例子中,官能化修饰的酶催化剂最高可给出87%的对映体过量值,而金属催化剂和有机小分子催化剂硫脲均只给出11%的对映体过量值。在硅正离子不对称抗衡阴离子导向催化这一思想的指导下,List课题组利用研发的亚氨基双膦酰亚胺酯(IDPi)成功解决了这一难题,通过其结合位点的受限空间的手性微环境,实现了前所未有的对映面细微识别(图1)。
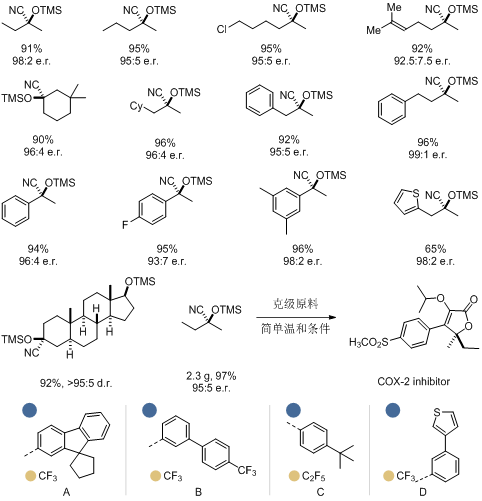
图2:底物拓展和产物应用(部分)(图片来源:Nature)
在具体的反应研究过程中,不管是脂肪酮,还是芳香酮均可以较好的对映选择性高效给出目标产物,相应的产物也已被证明在药物合成、农药研究中有较高的应用潜力(图2)。机理研究方面,核磁研究捕捉到在反应的初始阶段烯醇硅醚的形成,并通过控制实验证实了烯醇硅醚作为反应中间体的猜测。
香港中文大学(深圳)成贵娟教授课题组通过密度泛函理论计算提出了异氢氰酸作为活性反应中间体的新机理。计算发现催化剂IDPi与TMSCN反应生成硅基化的活性催化剂,并释放异氢氰酸。值得一提的是异氢氰酸的生成比氢氰酸更容易,并且其反应活性更高,异氢氰酸在催化剂的作用下与氢氰酸存在动态平衡。硅基化的催化剂对原料酮进行活化,生成氧碳鎓离子对中间体;该中间体接受异氢氰酸的进攻,生成目标产物硅基化氰醇并再生催化剂。此外,计算表明烯醇硅醚中间体是动力学产物,该中间体可以转化为热力学更稳定的氰基硅烷化产物(图3a)。详细的控制实验和核磁研究也为机理的阐述提供了事实依据。结构分析显示IDPi形成了包埋率高达73.4%的紧密结构,其中心骨架和取代基组成狭窄、不对称的反应口袋,区分生成两种对映异构体产物的过渡态结构(图3b)。计算表明催化剂的取代基可以调控催化剂反应口袋的形状、尺寸、包埋体积和位阻分布,从而形成不同的手性口袋来特异性识别脂肪酮和芳香酮等不同类型的底物。

图3:理论计算研究(图片来源:Nature)
(来源:科学网)
相关论文信息:https://doi.org/10.1038/s41586-022-04531-5









