|
|
| 何爱彬研究组开发CoTECH实现单细胞多维表观重构 |
|
细胞状态和细胞命运决定的机制一直是生物学领域普遍关心的基本问题。已有多种单细胞表观组技术被开发出来,如scChIP-seq1-5、scATAC-seq6等,用于解析命运相关决定过程中基因差异表达的调控机制。然而单组学的解析只能反映单个调控层次的信息,多组学解析可以进一步探究多个调控层次之间的互作,进而发现新的调控规律。已经有许多研究实现了在同一个细胞中进行多组学的解析,sci-CAR7、SNARE-seq8、Paired-seq9等能够在同一个细胞中捕获染色质开放程度和基因表达信息。但是由于scChIP-seq技术的难度与复杂程度,目前还很难实现在同一个细胞中高效地、高质量地、高通量地捕获蛋白质-染色质互作和基因表达信息。
2021年5月6日,北京大学分子医学研究所,北京大学-清华大学 联合中心何爱彬研究组在Nature Methods杂志在线发表题为“Single-cell joint detection of chromatin occupancy and transcriptome enables higher-dimensional epigenomic reconstructions”的研究论文,报道了一种新型单细胞多组学技术CoTECH (combined assay of transcriptome and enriched chromatin binding),该技术能够高通量地在单个细胞中同时捕获蛋白质-染色质互作和基因表达信息,并重构单细胞多维表观景图。
CoTECH技术在何爱彬课题组早期开发的scChIP-seq技术CoBATCH2的基础上引入了基于组合标签的单细胞mRNA捕获技术,实现了对单细胞转录组与表观组的高通量解析 (图1)。正是由于组合标签策略的沿用,CoTECH技术可以在不依赖微流控等特殊仪器的条件下高通量地对单细胞进行解析,即使在起始细胞数量有限(数千个单细胞)的情况下依然能获得很高的数据质量,能够满足大多数的研究需求。同时CoTECH具有极好的数据质量,可以在每个单细胞中捕获约3,000个非重复DNA片段与近8,000个UMI。
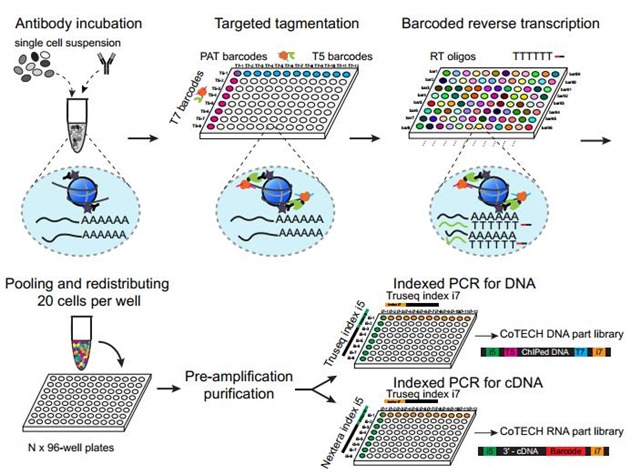
图1 高通量单细胞多组学技术CoTECH流程图
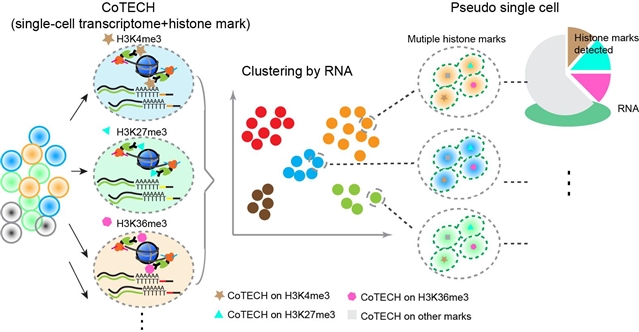
图2 CoTECH实现多种组蛋白修饰表观重构的分析策略
CoTECH技术的另一大特点是可以重构单细胞高维表观图景 (图2)。为了探究两种甚至多种组蛋白修饰的动态变化,作者根据CoTECH技术可以同时检测单细胞基因表达和组蛋白修饰的特性,提出一种全新的分析框架和策略,将若干基因表达相似的单细胞合并,通过设置“pseudosingle cells”,来分析对应的组蛋白修饰的特征。同时,以基因表达为媒介,可将多种不同的组蛋白修饰统一和联系起来,并实现多种组蛋白修饰的关联比较与整合分析。
二价染色质区域(bivalent chromatin domain)是指同时被H3K4me3与H3K27me3标记的染色质区域,与细胞分化过程密切相关。之前已有许多研究利用re-ChIP、质谱、成像等手段对二价性染色质的存在与分布规律进行探究,但目前还没有技术手段能够以单细胞分辨率对该区域进行全基因组无偏解析。利用CoTECH技术与“pseudosingle cells”的分析策略,研究者在小鼠胚胎干细胞中整合H3K4me3与H3K27me3的信息,模拟单细胞中二价性染色质区域的调控状态。研究者利用打分的方式对基因组区域二价性质的强弱进行量化,解析了随时序发展过程二价性染色质的动态调控图景。
小鼠造血系统的发育一共会经历三波造血,均涉及内皮细胞向生血内皮分化的过程,但是只有第三波造血过程会产生HSC,而前两波造血只能产生红系和髓系的祖细胞10。为了解析造血过程细胞命运决定差异化的机制,研究者收集E9.5 卵黄囊 (yolk-sac) 区域 (第二波造血) 及E10.5 主动脉-性腺-中肾 (aorta-gonad-mesonephros, AGM) 区域 (第三波造血) CD31+内皮细胞,并进行了H3K27ac (标记活跃增强子) CoTECH的解析。分析发现,在这两波造血过程中卵黄囊区域调控基因表达的增强子数目显著高于主动脉-性腺-中肾区域,并且具有分化偏好性基因的上游增强子对启动子的调控强度也具有偏好性。这些机制在一定程度上可以解释这两波造血过程中细胞命运产生差异的原因。
北京大学-清华大学 联合中心博士生熊海清、罗颖洁与北京大学分子医学研究所博士生王千昊为论文共同第一作者,北京大学分子医学研究所,北京大学-清华大学 联合中心何爱彬研究员为本文的通讯作者。该研究获得了科技部干细胞专项、国家自然科学基金委, 联合中心和勃林格殷格翰研究员奖项目的支持。(来源:科学网)
相关论文信息:https://doi.org/10.1038/s41592-021-01129-z
参考文献:
1. Rotem, A. et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 33, 1165-1172, doi:10.1038/nbt.3383 (2015).
2. Wang, Q. et al. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol Cell 76, 206-216 e207, doi:10.1016/j.molcel.2019.07.015 (2019).
3. Ai, S. et al. Profiling chromatin states using single-cell itChIP-seq. Nat Cell Biol 21, 1164-1172, doi:10.1038/s41556-019-0383-5 (2019).
4. Ku, W. L. et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat Methods 16, 323-325, doi:10.1038/s41592-019-0361-7 (2019).
5. Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930, doi:10.1038/s41467-019-09982-5 (2019)
6. Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486-490, doi:10.1038/nature14590 (2015).
7. Cao, J. et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380-1385, doi:10.1126/science.aau0730 (2018).
8. Chen, S., Lake, B. B. & Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol 37, 1452-1457, doi:10.1038/s41587-019-0290-0 (2019).
9. Zhu, C. et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol 26, 1063-1070, doi:10.1038/s41594-019-0323-x (2019).
10. Dzierzak, E. & Bigas, A. Blood Development: Hematopoietic Stem Cell Dependence and Independence. Cell Stem Cell 22, 639-651, doi:10.1016/j.stem.2018.04.015 (2018).








