|
|
|
|
|
大豆驯化过程中种间杂交对遗传瓶颈的消减作用 | Genome Biology |
|
|
论文标题:Genomic introgression through interspecific hybridization counteracts genetic bottleneck during soybean domestication
期刊:Genome Biology
作者:Xutong Wang, Liyang Chen and Jianxin Ma
发表时间:2019/01/30
数字识别码:10.1186/s13059-019-1631-5
原文链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1631-5?utm_source=other&utm_medium=other&utm_content=null&utm_campaign=BSCN_2_DD_GenBio_Arti_Scinet
微信链接:https://mp.weixin.qq.com/s/lOtQ20mXFMsP0bte_NyP0g
大豆(Glycine max [L.] Merr.)是重要的粮油兼用作物,是人类生活中重要的植物蛋白质来源。目前普遍认为栽培大豆起源于中国,在距今约6000至9000年前由野生大豆 (Glycine soja)驯化而来。这种单一起源的观点也被近来鉴定的驯化基因所支持 [1, 2]。尽管如此,大豆驯化的历史和过程至今仍充满争论。根据目前大豆基因组的测序结果估算,野生大豆与栽培品种间的分化程度达到距今约27万年的时间框,因而野生大豆发展到栽培大豆被认为是一个渐进的过程。在中国的大豆种植区,仍存在着形态介于野生大豆与驯化大豆间的半野生型大豆(Glycine gracilis)。
据此推测,可能存在野生大豆到栽培大豆的基因流(Gene flow)。
基因组片段渗入(Genomic Introgression)导致基因库间的大规模的基因流动,经种间杂交产生,为基因流(Gene Flow)的重要成因。基因渗入现象在玉米和水稻等作物中均有报道,然而对于作物驯化过程中基因渗入的进化模式及其所带来的进化结果均没有深入研究。近日,美国普渡大学马渐新教授团队利用他人发表的大豆种质的重测序数据 [3],通过比较与进化基因组的手段,揭示野生大豆和栽培大豆间细胞核及叶绿体的基因组片段及基因渗入模式,成因及动态进化结果。
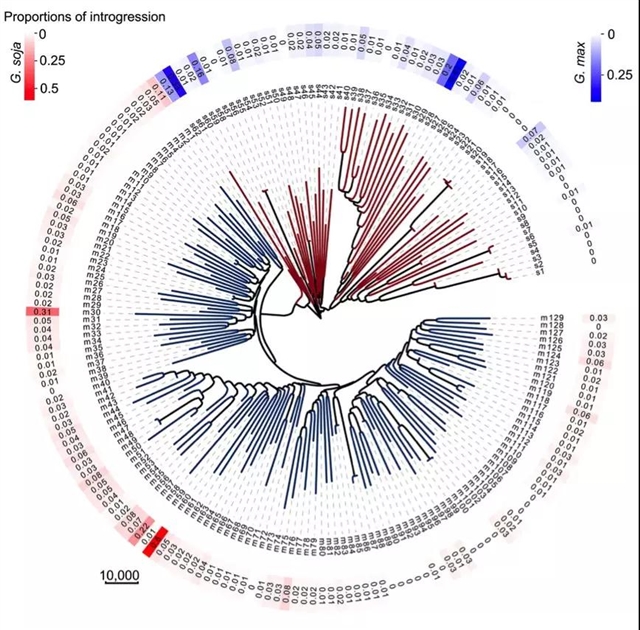
图1. 野生大豆和栽培大豆的进化树(Neighbor-joining)。
该研究通过对302份大豆种质的同源遗传关系分析 (Identical By Descent,IBD) [4], 发现几乎所有的大豆材料中存在种间基因渗入片段。该群体中的62份野生大豆材料和129份农家种未经历人工介导的有性杂交,所以每份材料中检测到的渗入片段的比例(图1)及这些片段在各条染色体分布情况(图2)应为大豆驯化过程中自然选择和人工选择的结果。该研究发现,在野生大豆和栽培大豆中近着丝粒区域的渗入片段比例要高于染色体臂,这可能与近着丝粒点区域的低重组率及对有害突变偏性积累有关。D-statics检验的结果表明这些渗入片段源自基因流(图3)而非消减系谱不完全分选效应(Incomplete lineage sorting)的结果。
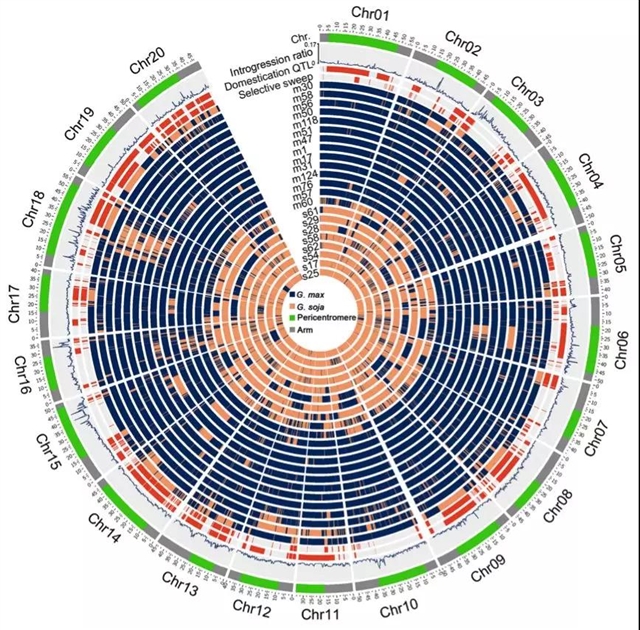
图2. 种间基因渗入在全基因组的分布。
环形从外至内依次表示 (i) 染色体臂(灰色),近着丝粒区(绿色);(ii) 群体水平的基因渗入比例在各条染色体的分布; (iii) 驯化相关QTL位点在各条染色体的分布(红色);(iv) 选择扫荡区域在各条染色体的分布(红色);(v) 具有代表性的12份栽培大豆及10份野生大豆材料基因渗入区间的在各条染色体的分布:栽培大豆片段(蓝色), 野生大豆片段(橙色)。
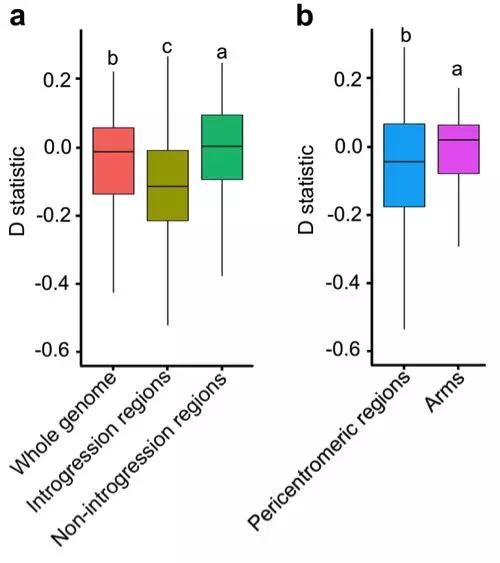
图3. D-statistic 分析方法展示野生和栽培大豆间基因组不同区域中不同模式的基因流。
该研究进一步对存在大片段基因组渗入的代表性材料进行了地理分布及全基因组及基因组局部区域的遗传多样性分析(图4)。其中野生大豆材料PI 578357(s61)和地方品种PI 393734(m30)中分别存在着33%和31%的渗入片段。s61来自黑龙江对岸的俄罗斯阿穆尔州,其二号染色体上的渗入区段与来自黑龙江省黑河地区的农家种 “黑河小黄豆”(m104)在进化树上存在极高的相似性,而s61中的非渗入区段则与另一来自俄罗斯滨海边疆地区的野生大豆材料s5存在极高相似性,同时这一材料在全基因组进化关系上与s61存在较大分歧。鉴于三个地区毗邻, s61中检测到的染色体嵌合(chimerism)有可能源自m30和s5(或与它们亲缘极近的材料)的种间杂交。同样,在采集于朝鲜半岛的地方品种m30的19号染色体中也发现了类似的现象,很可能是位于朝鲜半岛的地方种m111与野生大豆s42杂交的结果。根据全基因组序列推断,s61与m104和m30与s42分化时间约为37万和27万年前,它们之间基因组片段的渗入的模式应该是大豆驯化过程中栽培种与野生种杂交的直接证据。
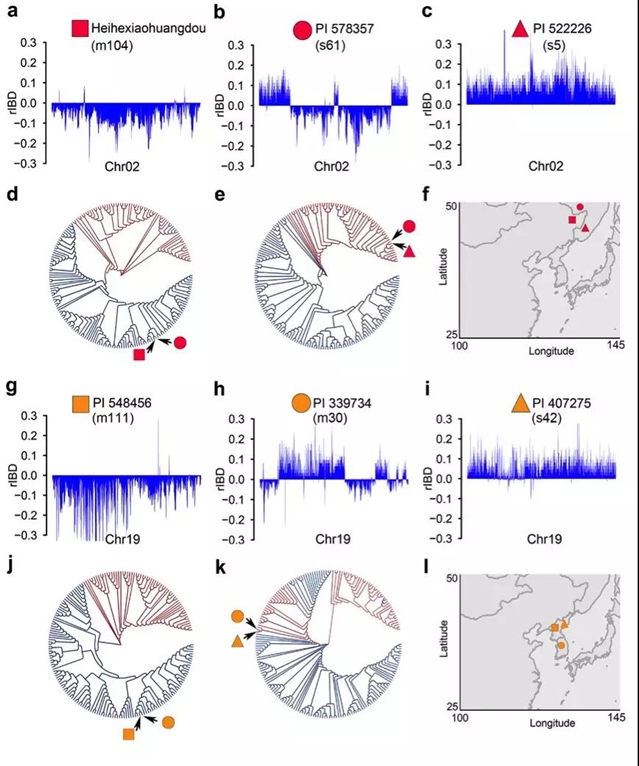
图4. 基于同源遗传关系分析推测的渗入片段起源的范例。
(a-c) 黑河小黄豆, PI 578357和PI 52226三个材料中基因组成分在2号染色体的分布。(d) 基于2号染色体基因渗入区域SNP构建的进化树揭示的PI 578357中PI 52226成分的起源。(e) 基于2号染色体非基因渗入区域SNP构建的进化树揭示的PI 578357中黑河小黄豆成分的起源。(f)上述三个材料的地理分布。(g-i) PI 548456, PI 339734和PI 407275三个材料中基因组成分在19号染色体的分布。(j) 基于19号染色体基因渗入区域SNP构建的进化树揭示的PI 339734中PI 548546成分的起源。(k) 基于19号染色体非基因渗入区域SNP构建的进化树揭示的PI 339734中PI 407275成分的起源。(l) 上述三个材料的地理分布。
理论上来说,野生种和栽培种间杂交与回交产生的后代材料要经历自然选择和人工选择: 前者选择对自然生存环境的适应性性状,而后者选择利于农业生产和人类消费的驯化性状。该研究揭示,野生品种中人工驯化选择性清除(selective sweep)区段栽培大豆渗入片段比例低于其他区段,而栽培品种中该区段野生大豆渗入片段比例也低于其他区段。在控制驯化性状的QTL区间 [5],上述渗入片段所占比例则更低(图5)。这些观察展示了自然选择和人工选择在栽培大豆和野生大豆杂交及回交后的不同选择效应。该研究还对驯化基因(GmHs1-1; B1)所在选择性清除[?](selective sweep)区段的SNP信息进行了分析,发现基因渗入现象在这两个区域均有存在(图 6)。
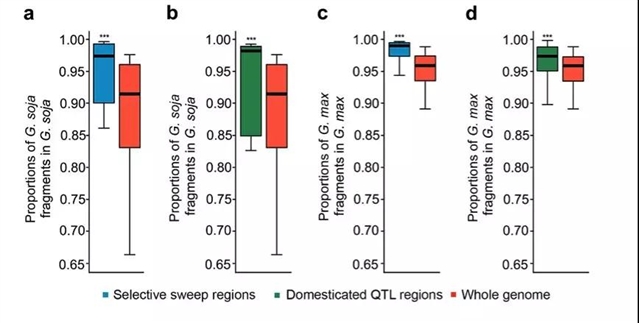
图5. 自然选择与人工选择下基因渗入的模式。
(a) 野生大豆中选择扫荡区段中野生大豆成分的比例。(b) 野生大豆中驯化相关QTL区段中野生大豆成分的比例。(c) 栽培大豆中选择扫荡区段中栽培大豆成分的比例。(d) 栽培大豆中驯化相关QTL区段中栽培大豆成分的比例。
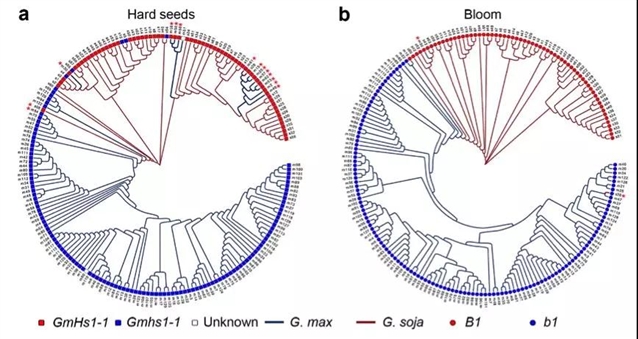
图6. 基于两个驯化相关基因所在的选择扫荡区段SNP信息构建的进化树。
该研究还探究了野生与栽培大豆中叶绿体基因组的进化关系。栽培大豆叶绿体的进化分化程度明显小于野生大豆,但有些栽培大豆中含有的是野生大豆类型的叶绿体基因组,有些野生大豆中检测到的是栽培大豆类型的叶绿体基因组(图 7)。这一观察提供了野生大豆与栽培大豆天然杂交的新证据。核基因组与叶绿体基因组总体上呈现出共进化趋势,但一些材料中细胞核与叶绿体基因组间发生了非平行进化,可能是种内天然杂交的结果。
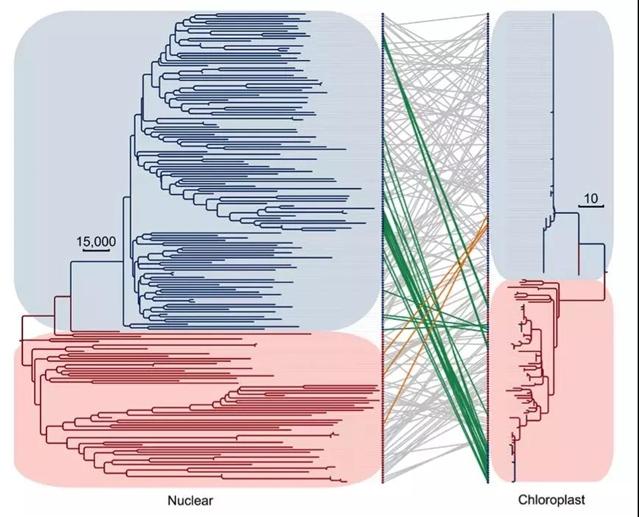
图7. 栽培大豆和野生大豆核基因组和叶绿体基因组的非对称分歧。
191个材料的核基因组进化树基于全基因组的SNP信息,叶绿体进化树基于333个高度可信SNP位点。栽培大豆材料用蓝色分枝蓝色背景表示,野生大豆材料用红色分枝红色背景表示。两个进化树中的相同材料用直线连接。蓝线表示栽培大豆材料中含有野生大豆叶绿体成分,橙线表示野生大豆材料中含有栽培大豆叶绿体成分。灰线表示材料的叶绿体成分来在亚群体内部。
作物驯化伴随着遗传多样性的大量丢失,是作物形成中的主要遗传瓶颈(Genetic Bottleneck)。但与野生大豆相比,栽培大豆中依然存在着较高的遗传多样性。据此估算,栽培大豆个体间的平均分化时间可追溯到30万年前(图 8),而这样高的多样性通常是无法通过对某个地域中少数野生材料的人工选择而获得的。该研究揭示了大豆驯化过程中种间杂交对遗传瓶颈的消减作用,对作物驯化过程的深入理解和种质资源的评价和利用具有指导意义。
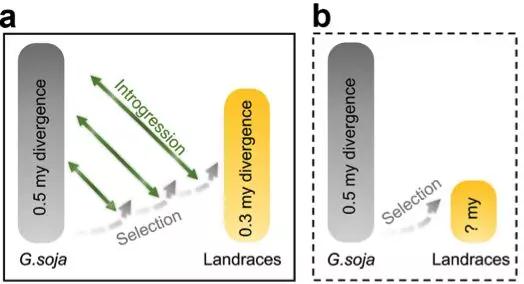
图8. 大豆驯化过程模型。
上述研究成果已于近日在Genome Biology在线发表,题目为 ”Genomic introgression through interspecific hybridization counteracts genetic bottleneck during soybean domestication”。美国普渡大学农学系及植物生物学中心马渐新教授为通讯作者,王旭彤博士为第一作者,博士研究生陈李洋为共同作者。
摘要:
Background
Evidence of introgression, the transfer of genetic material, between crops and their wild relatives through spontaneous hybridization and subsequent backcrossing has been documented; however, the evolutionary patterns and consequences of introgression and its influence on the processes of crop domestication and varietal diversification are poorly understood.
Results
We investigate the genomic landscape and evolution of putative crop-wild-relative introgression by analyzing the nuclear and chloroplast genomes from a panel of wild (Glycine soja) and domesticated (Glycine max) soybeans. Our data suggest that naturally occurring introgression between wild and domesticated soybeans was widespread and that introgressed variation in both wild and domesticated soybeans was selected against throughout the genomes and preferentially removed from the genomic regions underlying selective sweeps and domestication quantitative trait locus (QTL). In both taxa, putative introgression was preferentially retained in recombination-repressed pericentromeric regions that exhibit lower gene densities, reflecting potential roles of recombination in purging introgression. Despite extensive removal of introgressed variation by recurrent selection for domestication-related QTL and associated genomic regions, spontaneous interspecific hybridization during soybean domestication appear to have contributed to a rapid varietal diversification with high levels of genetic diversity and asymmetric evolution between the nuclear and chloroplast genomes.
Conclusions
This work reveals the evolutionary forces, patterns, and consequences of putative genomic introgression between crops and their wild relatives, and the effects of introgression on the processes of crop domestication and varietal diversification. We envision that interspecific introgression serves as an important mechanism for counteracting the reduction of genetic diversity in domesticated crops, particularly the ones under single domestication.
阅读论文全文请访问:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1631-5?utm_source=other&utm_medium=other&utm_content=null&utm_campaign=BSCN_2_DD_GenBio_Arti_Scinet
期刊介绍:
Genome Biology (https://genomebiology.biomedcentral.com/,13.2 - 2-year Impact Factor, 16.5 - 5-year Impact Factor) publishes outstanding research in all areas of biology and biomedicine studied from a genomic and post-genomic perspective.
(来源:科学网)
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






