|
|
| QB | 一种ChIP-seq弥散信号检测的新算法-RECOGNICER |
|
论文标题:RECOGNICER: A coarse-graining approach for identifying broad domains from ChIP-seq data (一种ChIP-seq弥散信号检测的新算法-RECOGNICER)
期刊:Quantitative Biology
作者:Chongzhi Zang(臧充之), Yiren Wang(王伊人), Weiqun Peng(彭卫群)
发表时间:19 November 2020
DOI:10.1007/s40484-020-0225-2
微信链接:点击此处阅读微信文章
真核细胞染色质内的组蛋白修饰(histone modification)是影响表观遗传状态和基因转录调控功能的重要因子之一。染色质免疫沉淀测序技术(ChIP-seq)自2007年面世以来,已成为测量染色质内各种蛋白质分子在全基因组内定位和分布的常用实验手段。与一般同特异DNA序列结合的转录因子(transcription factor)不同,组蛋白修饰的标记以缠绕着146bp DNA的核小体为单元,在基因组上定位的分辨率达不到单个bp那样精准,而且经常会标记在连续多个核小体上。组蛋白修饰的这些特点,在ChIP-seq实验数据中,就表现为基因组上的弥散信号,例如标记在异染色质的H3K9me3、体细胞的H3K27me3等,ChIP-seq数据上几乎不出现“尖峰”(sharp peak),给传统的“寻峰”(peak calling)算法带来挑战。同时,跟染色质三维空间构象的跨尺度分形特性相关,组蛋白修饰标记的基因组区域也存在几千bp到上百万bp跨越几个数量级尺度的特点。针对这些特征进行ChIP-seq多尺度“宽峰”检测,对原则性的生物信息学方法还存在需求。
近期,来自美国弗吉尼亚大学的臧充之、王伊人和乔治华盛顿大学彭卫群教授在Quantitative Biology上发表了题为“RECOGNICER: A coarse-graining approach for identifying broad domains from ChIP-seq data”的文章,介绍了一种新的ChIP-seq数据分析和弥散信号识别的生物信息学算法,即RECOGNICER(Recursive coarse-graining identification forChIP-seqenriched regions)。文章主要作者曾于2009年开发的SICER算法【1】是ChIP-seq弥散信号分析和检测组蛋白修饰类“宽峰”(broad peak)的有效生物信息学工具。在SICER基础上,作者在RECOGNICER中使用新模型识别ChIP-seq信号的多尺度聚集,适用于寻找基因组上更宽的信号富集区域。目前,RECOGNICER算法的代码全部开源(https://github.com/zanglab/recognicer),并已整合入SICER2软件包中(https://zanglab.github.io/SICER2/),希望可以成为ChIP-seq数据分析领域一个有用的生物信息学工具。
文章概要
RECOGNICER算法的原理来自于理论物理中重整化群的概念,利用粗粒化方法(coarse-graining)实现多尺度下的转换和计算。在操作中,该算法设计了一种区块变换(block transformation),自动处理在不同尺度下的信号聚集并递归,从而实现多尺度ChIP-seq信号富集区域的识别和统计分析(Figure 1)。
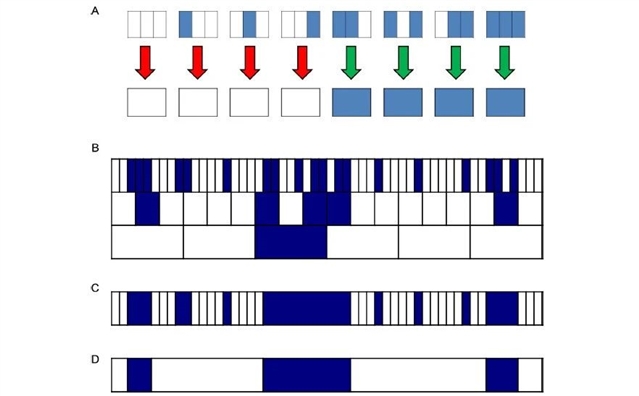
Figure 1. The RECOGNICER method: coarse-graining schematic. (A) Block transformation: The state of a block on the coarse scale is determined by its corresponding blocks on the fine scale according to the simplest majority rule (3 choose 2). Blue indicates blocks designated as “1”; white indicates blocks designated as “0”. (B–D) Analysis procedure: (B) Coarse-graining by recursive block transformation; (C) Domain retrieval to identify candidate regions on every scale; (D) Domain significance determination.
本文以H3K27me3 ChIP-seq公共数据为例,验证了RECOGNICER的性能。根据H3K27me3标记在沉默基因全区域的特点,在ENCODE协作组发布的多种细胞系ChIP-seq数据中,RECOGNICER能够在更多的沉默基因上检测到被一个完整的H3K27me3宽峰覆盖,而不是断续的多峰,这一结果优于现有的几种ChIP-seq宽峰检测工具(Figure 2)。
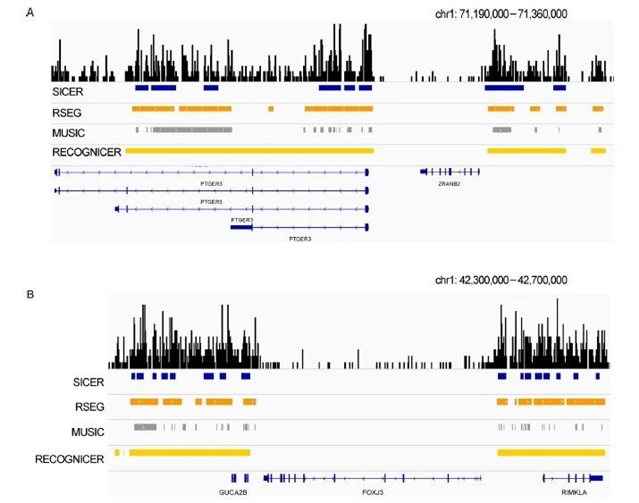
Figure 2. Examples of H3K27me3 board domains identified using different tools. (A) H3K27me3 marks the silent gene PTGER3 (left) while an active gene ZRANB2 (right) is not marked. (B) Two H3K27me3 broad domains are bounded at chromatin regions flanking an active gene FOXJ3.
Reference
1. Zang,Chongzhi; Schones, Dustin E.; Zeng, Chen; et al. A clustering approach foridentification of enriched domains from histone modification ChIP-Seq data. Bioinformatics,2009, 25(15): 1952-1958
摘要:
Background: Histone modifications are major factors that define chromatin states and have functions in regulating gene expression in eukaryotic cells. Chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) technique has been widely used for profiling the genome-wide distribution of chromatin-associating protein factors. Some histone modifications, such as H3K27me3 and H3K9me3, usually mark broad domains in the genome ranging from kilobases (kb) to megabases (Mb) long, resulting in diffuse patterns in the ChIP-seq data that are challenging for signal separation. While most existing ChIP-seq peak-calling algorithms are based on local statistical models without account of multi-scale features, a principled method to identify scale-free board domains has been lacking.
Methods: Here we present RECOGNICER (Recursive coarse-graining identification for ChIP-seq enriched regions), a computational method for identifying ChIP-seq enriched domains on a large range of scales. The algorithm is based on a coarse-graining approach, which uses recursive block transformations to determine spatial clustering of local enriched elements across multiple length scales.
Quantitative Biology期刊介绍
Quantitative Biology (QB)期刊是由清华大学、北京大学、高教出版社联合创办的全英文学术期刊。QB主要刊登生物信息学、计算生物学、系统生物学、理论生物学和合成生物学的最新研究成果和前沿进展,并为 与计算机、数学、物理等交叉研究领域打造一个学术水平高、可读性强、具有全球影响力的交叉学科期刊品牌。

特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。








