|
|
|
|
|
基于“1,4-镍迁移”策略实现芳基溴化物的原位/邻位双官能化 |
|
|
近日,南京大学朱少林课题组和西班牙加泰罗尼亚化学研究所(ICIQ)Ruben Martin课题组合作,基于“1,4-镍迁移”策略实现芳基溴化物的原位/邻位双官能化反应。
2021年11月18日,该研究以“Nickel-Catalyzed Ipso/Ortho Difunctionalization of Aryl Bromides with Alkynes and Alkyl Bromides via a Vinyl-to-Aryl 1,4-Hydride Shift”,发表在J. Am. Chem. Soc.上。南京大学特任副研究员何玉立博士为论文第一作者。
多取代芳烃是药物分子和功能材料的重要骨架,从卤代芳烃出发,目前合成这类化合物的主要方法有缺电子芳环的亲核取代反应或交叉偶联反应,但是上述方法局限于原位发生官能团化一次只能引入一个取代基(如图1,方法a);钯催化的Catellani类型反应为多取代芳烃的合成提供了高效的方法,在芳基卤化物的原位和邻位同时引入官能团得到一部反应引入两个取代基的多取代芳烃,然而这类反应一般需要当量的降冰片烯参与,并且常规的反应条件为了避免二次邻位官能团化,要求参与反应的卤代芳烃底物的邻位必须预先放置一个取代基,限制了反应的底物范围(如图1,方法b)。因此开发廉价金属催化的通用性强、选择性高的模块化合成多取代芳烃的新方法是科学家们一直追求的目标。
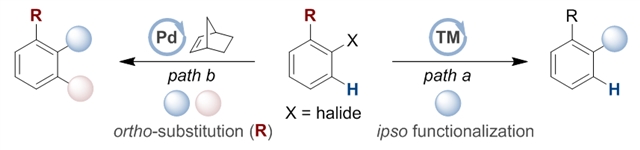
图1:芳基卤化物的交叉偶联
南京大学朱少林课题组前期利用镍在脂肪碳链上的“1,2-镍迁移”策略实现了远程sp3C-H的多种官能团转化(如图2,左上)。而镍在芳香碳链上的“镍迁移”却鲜有报道,仅有的一例反应为2020年西班牙加泰罗利亚化学所(ICIQ)Ruben Martin 教授等人利用“1,4-镍迁移”策略实现了远程sp2C-H的羧基化反应(如图2,右上)。受此工作启发,朱少林团队和Ruben Martin课题组合作,成功实现了更具挑战性的芳基溴、烷基溴和炔烃的多组份模块化的迁移偶联反应进而构建出一系列的多取代芳烃。
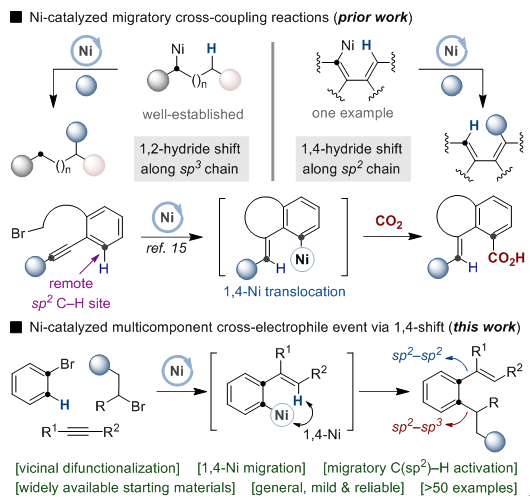
图2:镍催化的迁移偶联化学
设想的反应基本过程如图3所示:首先零价镍(I)与芳基溴(1)氧化加成,接着与炔烃(2)发生顺式迁移插入反应得到烯基镍中间体(III),继而进行“1,4-镍迁移”得到芳基镍物种(IV),然后捕获通过单电子转移(SET)产生的烷基自由基(V)得到三价镍中间体(VI),最后还原消除生成双官能团化产物(4)和一价镍(VII),VII同时可以单电子还原烷基卤代烃(3)得到二价镍(VIII),在锰粉的作用下的,VIII最终还原成零价镍(I),完成催化循环。
模板反应可以在最优条件下,以74%的分离产率、96:4的Z/E选择性和96:4的区域选择性得到目标产物。从条件筛选表格中我们可以看到联吡啶配体6位的甲基取代在反应中起到至关重要的作用,同样的,添加剂碘盐和4 Å分子筛也影响反应的产率和选择性。
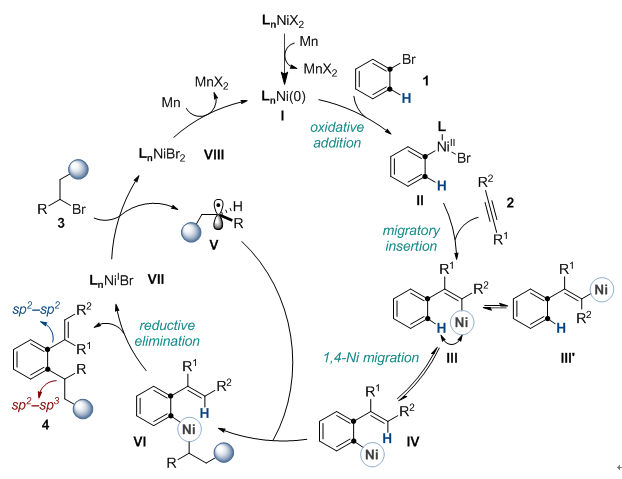
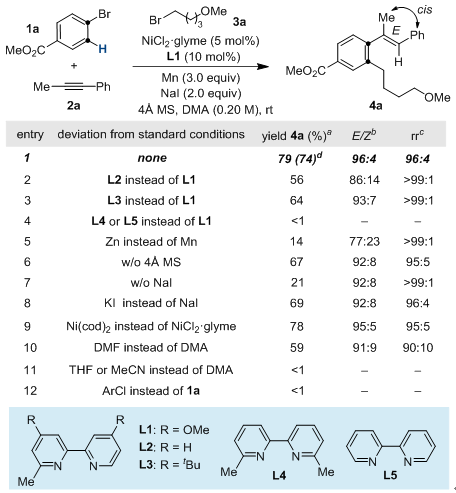
图3:反应条件筛选
作者在最优条件下对该三组份反应进行了底物范围的考察。芳基溴上无论是缺电子和富电子的取代基均能给出较好的产率和优秀的选择性,值得注意的是,当使用非对称的芳基溴时,“1,4-镍迁移”则发生在位阻更小的邻位。芳基和烷基取代的炔烃也能得到令人满意的结果。一级和二级烷基溴化物同样适用于该体系,且可以用于多种药物活性分子的后期修饰。
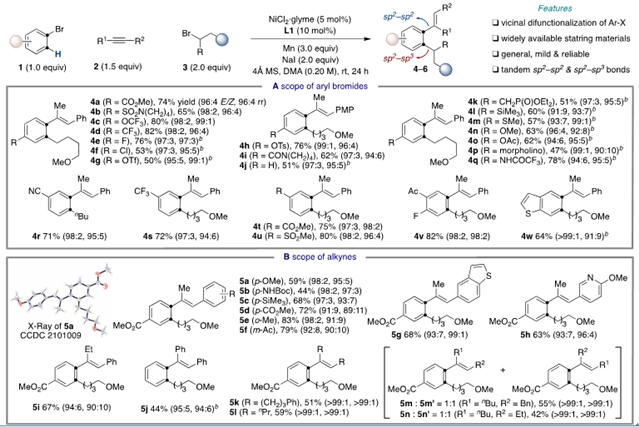
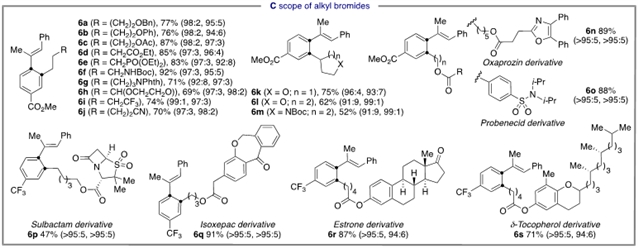
图4:镍催化三组份迁移烷基化反应
作者随后对目标产物进行了多种转化,如钌催化的氧化、钯催化的氢化反应,进一步证明了该体系可以从商业易得的原料制备多取代芳烃的实用性和可行性。
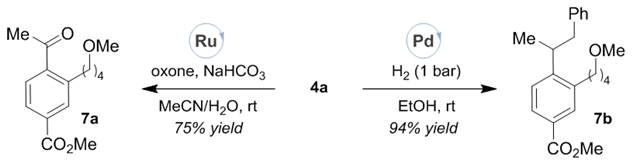
图5:合成应用
为验证反应机理,作者进行了一系列同位素标记实验,当使用d5-溴苯作为反应底物时,可以发现芳基上的氘可以全部迁移到烯基碳上,间接证明了“1,4-镍迁移”发生在烷基化之前,动力学同位素实验表明“1,4-镍迁移”应该不是反应的决速步。根据机理推测中的中间体III,作者认为反应同样可以从烯基溴出发进一步发生分子内的“1,4-镍迁移”,因此作者使用(Z)和(E)两种构型的烯基溴化物,在最优条件下均能得到同样构型和选择性的目标产物,这也说明在发生迁移之前反应中间体III的顺反构型可以快速翻转。
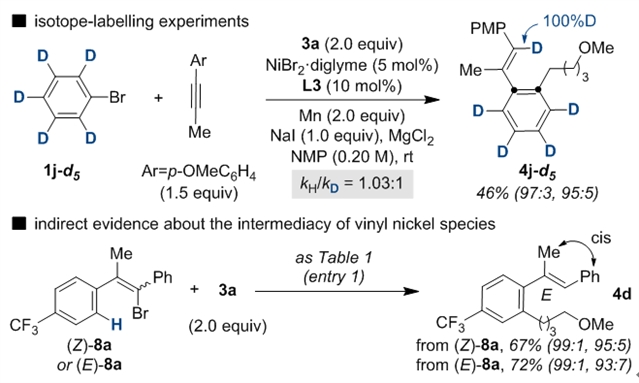
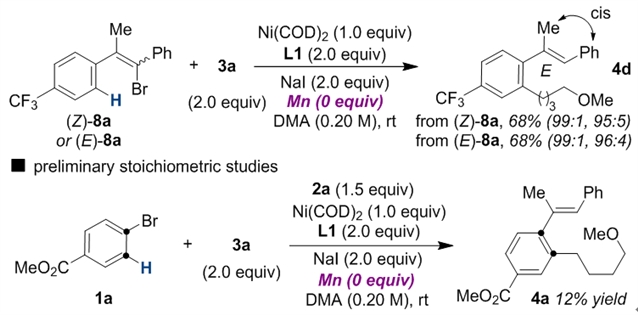
图6:机理实验研究
南京大学朱少林课题组和西班牙加泰罗尼亚化学研究所(ICIQ)Ruben Martin课题组合作,通过“1,4-镍迁移”策略实现芳基溴化物的原位和邻位双官能化,为多取代芳烃的合成提供新思路,同时也开辟了镍催化迁移化学的新方向。(来源:科学网)
相关论文信息:https://doi.org/10.1021/jacs.1c10368






