“诺奖之后,AI再立新功!”
10月9日,就在2024年诺贝尔化学奖颁给“蛋白质结构预测”的当天,阿里云和中山大学也收到好消息:他们联手用人工智能(AI)发现16万余种RNA“暗物质病毒”的论文,在国际顶级学术期刊《细胞》(Cell)上发表了。
之所以将新发现的RNA病毒称作“暗物质病毒”,论文共同通讯作者、中山大学教授施莽告诉《中国科学报》,新发现的这16万余种RNA病毒与已知病毒“不具备同源性或者只有极低同源性”,如果不是借助AI技术手段,使用过去常用的“基于序列同源性比对”的生物学方法很难发现它们。
“新方法能够更快、更准地判别未知的RNA病毒,有望改变病毒发现的研究范式,改变人们对RNA病毒多样性和病毒演化历史的认知。”施莽说。
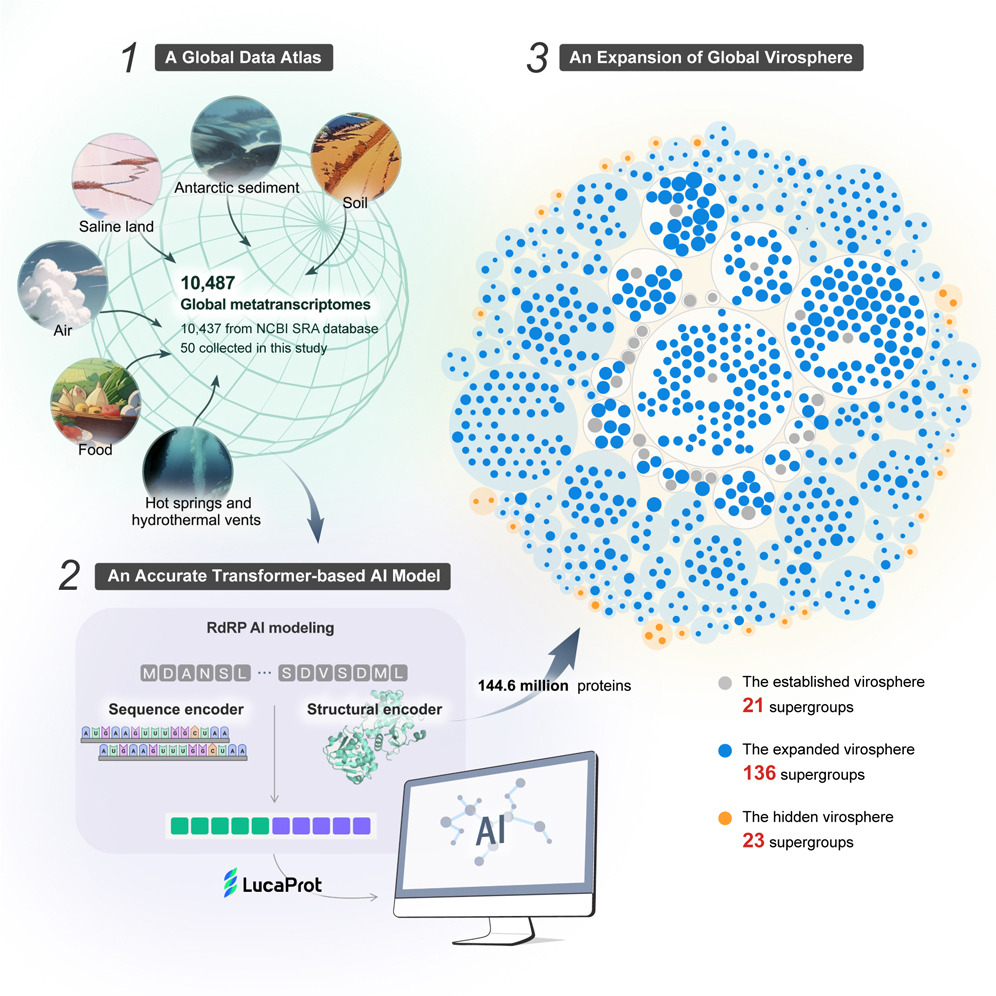 用AI发现RNA新病毒论文图示摘要。图源:Cell
用AI发现RNA新病毒论文图示摘要。图源:Cell
?
打破病毒发现瓶颈,AI再立新功
发现新病毒是病毒学研究的基础工作之一。在地球生态系统和人类进化史上,病毒扮演的角色可谓举足轻重;然而,直至今日人类对病毒的了解仍知之甚少,目前已知的病毒种类也只是病毒世界的冰山一角。因此,用更高效、更精准的方法发现新的病毒是研究者一直以来的追求。
然而,使用现有的生物信息学方法,未知病毒的发现工作似乎陷入停滞,难以取得更进一步的突破。尽管坚信“还有更多未知的病毒类型等待我们去发现”,施莽在联手阿里云开展这项研究之前,心里也不停在打鼓:“病毒组的探索是否已经进入了瓶颈期?”
一次交流的机会,施莽与阿里云飞天实验室生物计算总监李兆融聊起了这个问题。后者提出,或许可以通过AI方法寻找突破口。听过了李兆融关于AI以及大模型技术的能力介绍和分析后,施莽立刻意识到:有戏!
两人一拍即合,一项围绕未知RNA病毒发现的合作项目在中山大学和阿里云之间旋即展开。
 合作研究团队合影(右二至右四分别为贺勇、李兆融、施莽)。贺勇 供图
合作研究团队合影(右二至右四分别为贺勇、李兆融、施莽)。贺勇 供图
?
在第一次会面后,论文共同一作者、阿里云算法专家贺勇在了解了链路长而繁琐的传统方法之后,认为更“端到端”的AI模型应对这一问题的效果一定会很好。
“我们的合作非常默契。”施莽介绍说,中山大学主要负责病毒学相关的问题,比如将收集的病毒相关大数据提供给阿里云,阿里云生物计算团队则负责利用这些数据,结合最先进的技术构建AI模型。在交流和完善方法的过程中,双方还“彼此科普对方不熟悉的领域”。
 贺勇向施莽课题组介绍Transformer架构。施莽 供图
贺勇向施莽课题组介绍Transformer架构。施莽 供图
?
具体来说,研究团队设计了基于Transformer架构的深度学习模型——LucaProt,该模型利用了对公开数据集的上亿蛋白质进行学习的大模型,融合原始序列和结构信息完成训练,是一种能进行蛋白质功能预测的深度学习网络。
这也正是这项研究最大的创新点。施莽告诉记者,和蛋白质类似,RNA这类生物大分子遵循一条基本法则:序列决定结构、结构决定功能。过往研究通常采用单一的序列信息用作AI建模,很容易遗漏那些序列相似度较低的多样化RNA病毒;而结构信息的引入,可以大大提升AI发现RNA病毒的速度和准确性。
随后,研究团队将LucaProt模型应用到海量新数据的预测中。
他们在全球范围内收集了10487份生物环境样本——它们来自南极底泥、深海热泉、活性污泥、盐碱滩等等,并将这些样本中所有微生物的RNA转录本结集整合,得到环境样本的微生物宏转录组。
有了这些环境样本的微生物宏转录组,LucaProt就有了用武之地——它通过挖掘这些环境样本中RNA病毒的唯一共有保守蛋白酶RdRP的基因序列,就能找到隐藏在这些环境样本中未知的RNA病毒。
最终,在这10487份环境样本中,AI共发掘出180个病毒超群(注:在病毒分类学中,病毒超群通常包含多个基因组类型相似的病毒家族或亚群)、161979种病毒。这一数量,直接将RNA病毒超群数量扩容约8倍、病毒种类扩充约30倍。
他们还发现其中一种病毒包含47250个核苷酸,是有史以来被报道的最大的RNA病毒。
新发现颠覆RNA“病毒圈”认知
为了排除“假阳性”的可能,研究团队还从序列同源性和结构同源性和现有的综合数据库比对,确定这些病毒和现有其他生物序列不具备相似性;同时,课题组还对从南极等地采集的50多个样本同时进行DNA和RNA测序,验证他们发现的RNA病毒不是来自于DNA生物的污染(注:用以判定RNA病毒的聚合酶RdRP的基因序列也存在于真核生物中)。此外,他们还进一步抽样做PCR实验验证。
“多个角度充分证明新发现的病毒结果真实可靠。”贺勇告诉《中国科学报》,结构信息的引入大幅提升了AI发现RNA病毒的速度和准确性:AI每发现一个病毒的平均耗时为几百毫秒到几秒,效率远高于经典方法所需的几天到几周;而多个来源的外部验证集上测试结果显示,反应模型正确预测的召回率高达97.4%,假阳性率仅0.023%。
这项研究几乎颠覆了人们对RNA“病毒圈”的认识。
贺勇对记者表示,此次发现16万余种RNA新病毒,绝大部分都是与已知病毒同源性很低的“暗物质病毒”,甚至有23个病毒超群是传统方法所不能发现的。
“这显示了人工智能模型在病毒学领域的重要意义。”他说。
更重要的是,据了解,此前,科学家发现RNA病毒的来源主要是人和动物等宿主,直到近年来才逐渐拓展到无脊椎动物和环境样本,但相关研究仍处于早期。这项研究是业界首次在环境样本中发现如此巨量的新病毒,大幅提升了学术界对病毒圈多样性的认知,有望为RNA病毒研究工作打开全新的空间。
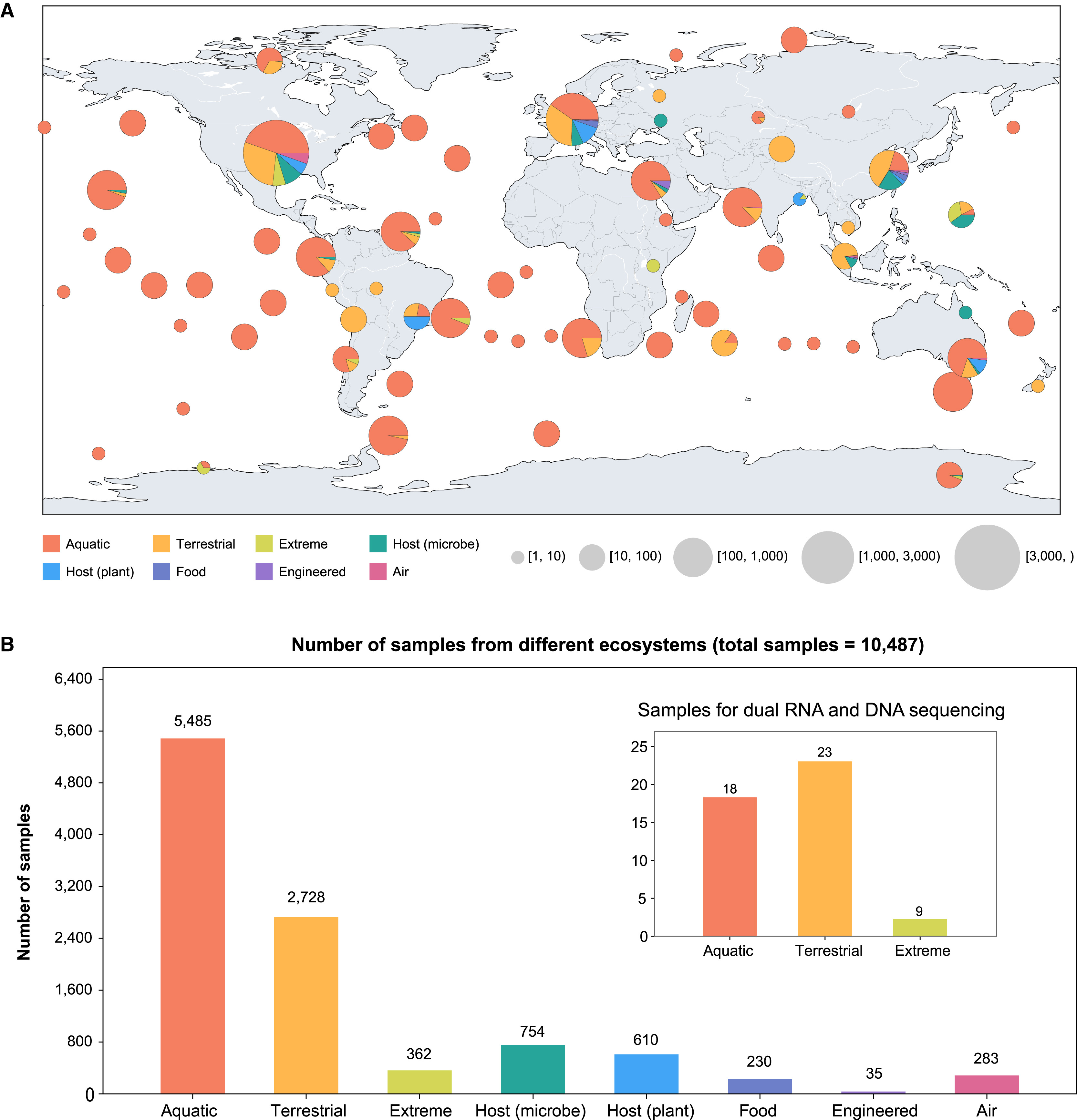 该研究中分析的宏转录组数据的地理覆盖范围。图源:Cell
该研究中分析的宏转录组数据的地理覆盖范围。图源:Cell
?
比如,研究团队通过对发现的RNA病毒来源分布进行分析的结果显示,尽管落叶层、湿地、淡水和废水环境的病毒多样性最高,但在极端环境中的RNA病毒多样性和丰度并不低,甚至在高温的深海热泉等对RNA不利的环境中,RNA病毒仍然在活跃复制。施莽认为,这说明RNA病毒不只是某个地方某种生物环境中存在,而是具有一定程度的广泛性。
“基于AI+病毒学的新研究框架刷新了人类对病毒圈的认识。”贺勇说,这种认识的不断完善,将有助于人类对未来可能发生的大流行进行预警,并进一步推动RNA病毒疫苗的研发。
AI for Science正引发科研范式深刻变革
连日来,2024年诺贝尔物理学奖、化学奖都授予了做人工智能相关研究的学者,引起了人们对于AI改变科学研究范式的广泛讨论。李兆融提出,中山大学和阿里云合作的这项研究,是AI for Science深刻变革的又一力证。
“这个框架正在逐步成为该领域的前沿工具,也开始被应用到其他类型的蛋白质鉴定和功能发现任务上。”李兆融说,该研究中的AI模型LucaProt现已开源,开发者和其他研究者可下载模型部署在本地或直接在线使用,也可对模型进行重新训练和调优,用于其他的蛋白质功能预测任务。
施莽认为,在科研领域,AI的应用已经势不可挡,通过AI方法探索科学问题已取得了重要突破。“这种研究范式将成为未来科学界的常态,也可能成为我们认知世界的重要手段。”
他告诉《中国科学报》,最近他参加了一次关于X病原体预警的大会,“人工智能”一词在至少3/4的报告中被提及、在1/4的报告中被用到。
“要知道,这次会议并非计算生物学和生物信息学领域。”施莽说,他因此认为,人工智能不仅改变了蛋白质科学,更重塑了整个 的研究范式。
近期,我国计算机科学家、中国工程院院士李国杰围绕《诺贝尔物理学和化学奖为何偏爱人工智能?》发表观点并撰文,他指出,这次诺贝尔奖的“出圈”不仅不是物理学界和化学界的“悲哀”,反而“可能在科学史上翻开新一页”。
在该文中,李国杰犀利地指出,科学语言和科学方法适合处理精确问题,具有局限性和相对性;随着一些简单的科学问题被解决掉,现在留下的都是“包含随机性和不确定性的复杂问题”,而人工智能更适合处理这种难以精确描述的问题。从某种意义上来讲,是“复杂性挑战逼迫人工智能和所有的传统科学形成了‘统一战线’”。
在AI发现RNA病毒这项研究中,这一论点得到了充分的支撑。贺勇提到,无论是模型训练规模上还是研究对象的数据维度上,这项研究呈现出了传统方法难以触及的复杂度,而自动化工具也展现了比传统生物信息学更高效、更全面的分析能力。
“这不仅对病毒学具有重要影响,还为生物医学领域的其他研究提供了新的视角。”贺勇说。
不过,研究团队也表示,这项研究只是深度学习在病毒发现领域的初步探索,相信今后会出现更多更高效、更准确的模型工具,不断迭代生信分析方法。相信借助更高效的工具,病毒学界将能在更大尺度上实现更高通量的病毒发现,并进一步提高病毒识别的精度。
相关论文信息及模型地址:
https://www.cell.com/cell/fulltext/S0092-8674(24)01085-7
https://github.com/alibaba/LucaProt
http://lucaprot.org/
版权声明:凡本网注明“来源:中国科学报、科学网、科学新闻杂志”的所有作品,网站转载,请在正文上方注明来源和作者,且不得对内容作实质性改动;微信公众号、头条号等新媒体平台,转载请联系授权。邮箱:shouquan@stimes.cn。






