|
|
| QB | 纪念HGP20周年系列文章9:哈佛大学John Quackenbush教授回顾基因组研究的前二十年 |
|
论文标题:Looking back at the first twenty years of genomics(哈佛大学John Quackenbush教授回顾基因组研究的前二十年)
期刊:Quantitative Biology
作者:John Quackenbush
发表时间:02 Mar 2022
DOI:10.15302/J-QB-021-0286
微信链接:点击此处阅读微信文章

2021年,QB期刊发表了一系列文章(文末附文章链接)庆祝第一张人类基因组草图发表20周年。2022年3月31日,Science 在线发表了端粒到端粒 (T2T) 联盟的“ The complete sequence of a human genome”研究论文。第二天,Science在线又发表了基于该完整的端粒到端粒人类基因组组装(T2T-CHM13)序列的另外五篇文章,分别从人类遗传变异分析(A complete reference genome improves analysis of human genetic variation)、人类片段重复组织的全面视图(Segmental duplications and their variation in a complete human genome)、人类着丝粒序列分析(Complete genomic and epigenetic maps of human centromeres)、人类参考基因组的从头重复发现和注释 (From telomere to telomere: The tranional and epigenetic state of human repeat elements)、完整人类基因组的表观模式(Epigenetic patterns in a complete human genome)这五个方面进行了详细阐述。回顾人类基因组学研究这二十年来的历程,科学家们一直在探索解码人类自身的奥秘。探索过程中不仅有重大突破带来的胜利喜悦,同时科学家们得到的经验和反思同样也是一笔宝贵的财富。
QB期刊邀请了美国Harvard T.H. Chan School of Public Health的John Quackenbush教授在今年最新一期的文章中分享了他的故事:Looking back at the first twenty years of genomics(点击文末“阅读原文”下载PDF全文)。

John Quackenbush 教授
Profile
John Quackenbush是哈佛大学陈曾熙公共卫生学院(Harvard T.H.Chan School of Public Health)生物统计系主任、计算生物学和生物信息学教授、网络医学部教授、Dana-Farber癌症研究所教授。他是理论物理学博士,1992年开始从事HGP研究,研究方向是计算生物学和生物信息学。目前,他已发表300多篇文章,总被引次数超过73000次。他的荣誉之一是在2013年被公认为白宫变革开放科学冠军(White House Open Science Champion of Change)。
文章概要
二十年前两个人类基因组几乎同时发表,当时“草图”的概念非常模糊,导致了很多怪事——2001年宣布草图完成,2006年宣布最长的1号染色体测序完成,而其它染色体完成时间还要更晚,比如X染色体在2020年才宣布完成,Y染色体至今没有消息。20年来人们为完善人类基因组做了大量工作,2021年,一份完整的人类基因组终于发表在bioRxiv上(注:文章已于2022年发表在Science上)。
目前关于基因组还有很多问题有待回答,比如人类基因的数量、基因的定义等等,我们目前得到的任何版本的基因组都仍然是不完美、不精确的,但是是非常有用的。回顾基因组研究这前二十年,John教授提出了“技术和数据是最重要的”,“生物系统的复杂性”,“开放式科学的重要性”和“提出没有答案的问题是有价值的”这四条宝贵的经验。
1. 技术和数据是最重要的
HGP刚开始时,所有人都意识到当时的测序技术是无法测完整个基因组的,因为测序成本过高。一般估计第一个人类基因组的成本在3亿到30亿美元之间,但由于有许多不得不尝试的技术路径,其最终成本可能更高。而第二个基因组的测序成本就降到了大约1亿美元,在之后的几年里,单位基因组的测序成本以大约每18到24个月减半的速度在下降,直到2007年降到了100万美元,今天这个数字大约是1000美元(图1)。
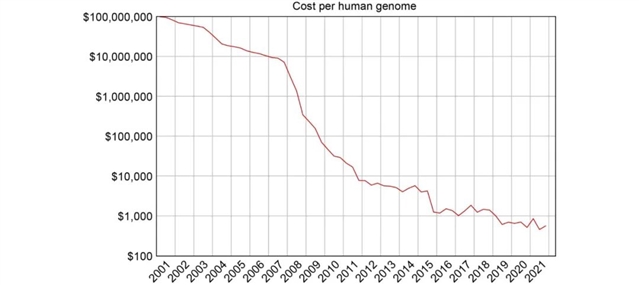
图1. 2001年至2021年,人类基因组测序成本的变化
测序成本的降低普及了基因组研究,许多学者在一般的医学研究里也会应用基因组技术。一些大项目,如Biobank,the Million Veterans Project,TOPMed和All of US提供了近一百万个完整的人类基因组,全基因组关联研究(GWASes)则经常在100万或更多的人群中对多达10,000,000个单核苷酸多态性(SNPs)进行基因分型。预计GWAS人群在许多项目中会增长到5,000,000个。如此规模的数据量则需要更好的数据管理技术和数据分析方法,通过比较大量的数据,人们才有可能从中发现规律。因此,我们看到健康和生物医学研究逐渐演变为技术驱动的数据和 研究,其中最有能力取得进展的是有能力收集、管理和分析大型复杂数据的方向。
2. 生物系统的复杂性
在HGP中有许多惊人的发现,其中之一是人类居然只有那么少的蛋白编码基因,仅有25000个左右,而一毫米长的线虫就有约20000个。当基因组草案公布时,许多人声称,拥有一个几乎完整的人类基因目录将使我们能够迅速发现大多数人类疾病的遗传根源,并确定许多性状的驱动因素。许多科学家开始相信,通过全基因组关联研究(GWAS),可以将包括遗传性疾病在内的常见性状与常见突变联系起来。低成本、高通量SNP基因分型阵列的发展,合理地全面覆盖了人类基因组中的常见突变,使GWAS和定量性状基因座(QTL)分析在经济上是可行的。实际上也确实可行,自2005年第一个GWAS研究以来,已经有近50000对SNP和复杂性状之间的关联被报告,这些研究也帮助发现了很多致病基因。
尽管取得了这些成功,但GWAS信号所解释的表型变异的百分比通常不高,而且大多数复杂性状的遗传性仍未得到解释。例如,对GWAS数据的综合荟萃分析发现,697个遗传变异可以解释人类身高的20%,但要将解释力提高到21%,需要近2000个,要达到29%的解释力,需要9500个。罕见的遗传变异,自发地出现在有限的家庭群体或个人中,被认为可能是疾病的驱动因素,但这些也被发现没有什么解释力。研究基因位点与基因表达关系的eQTL也已经发现了数万个,但很少有与表型和疾病直接产生因果关系。
这些失败表明,生物状态是由复杂的相互作用的细胞元素网络定义的,它们共同作用改变细胞过程,并最终改变表型状态。决定表型的不是单个SNP,而是以非线性方式改变生物功能的变异家族。这个概念类似于后来提出的 " omnigenic ",它表明个体的遗传背景会微妙地改变单个细胞的调控途径和基因表达,最终促成表型的形成。一些模型对这一过程进行了建模,如PANDA,LIONESS等。这些复杂的网络分析发现了与生物相关的调节过程的变化,即使在基因表达很少有明显差异的情况下也能区分表型。这为分析和模拟生物过程开辟了新的途径,提供了估计和比较不同条件下的调控网络的方法,人们甚至认识到网络的差异可能比用来构建网络的组学特征更有作为生物标志物的价值(图2)。鉴于基因数量相对较少,它们不仅有助于解释人类表型的复杂性,而且可以帮助理解细胞过程对扰动的稳定性,以及发育和进化过程中的复杂变化。当然,如果没有人类基因组计划,这一切都不可能发生。

图2. 许多组学数据的分析依赖于不同表型之间的组学特征比较
3. 开放式科学的重要性
当人类基因组测序开始的时候,这个项目对于任何一个单一的学术机构来说都过于庞大和复杂。公共基因组计划在很大程度上是围绕着 "分而治之 "的方法进行的,各种染色体或染色体区域分配给各个研究组或财团,他们能够产生连续的测序数据并发表他们的分析结果。但资助者和组织者也认识到,最有用的基因组序列应该符合最低质量标准,并且能够提供免费和开放的基础数据。因此1996年在百慕大召开的会议达成了一项生物医学研究中前所未有的协议——要求所有DNA序列数据在生成后24小时内公布在可公开访问的数据库中,而不是等待发表。这重申了研究中的一个基本原则——任何科学调查的结果都应该开放给其他科学家重新分析和验证, 在测序领域这个原则就是要提供数据。虽然公共基因组在重分析中暴露了许多问题,但作者认为这恰恰是公共工程的优越性,因为数据的可用性意味着人们有机会发现错误,而Celera公司的私有基因组虽然也可以下载,但他们限制了每个研究组的下载速度,以至于下载他们的基因组将花费一位科学家大约58年时间。
这种对基本科学原则和开放科学的承诺在整个基因组学界产生了反响。许多大规模基因表达分析的先驱者成立了微阵列基因表达数据协会,并积极创建了开放数据的微阵列实验最低信息(MIAME)标准,然后不懈地推动期刊、资助者和科学家将“共享有用的数据”作为出版和资助的要求。过去20年里,从基因组测序到单细胞分析、代谢组学、蛋白质组学、体内研究,甚至医学人工智能报告等领域都建立了30多个数据共享的最低信息标准。
数据共享发现了许多问题,如Anil Potti领导的科学家团队发表了一系列论文,声称发现了可以预测病人对化疗反应的基因表达特征,他们启动了三项临床试验并成立了一家公司。但是包括MD安德森癌症中心的Keith Baggerly在内的一些科学家无法独立复制Potti的结果,并发现发表的数据与作为研究数据的内容之间存在矛盾。最终纸牌屋倒塌了,试验停止,论文被撤回,Potti也离开了学术界。负责调查该事件的一个委员会呼吁, 任何旨在用于临床应用的研究,不仅要公布数据,还要公布方法和开源软件(图3)。
图3. 美国科学院发布的“Evolution of Translational Omics: Lessons Learned and the Path Forward”报告
开放科学(包括共享数据、模型和软件)的重要性正在被基因组学界以外的人所认可,特别是在为临床应用而开发的检测中,这一点至关重要。当谷歌公司的数据科学家领导的团队发布了一个乳腺癌放射学分类系统,声称它足够强大,可以开始临床试验时,参与确保基因组生物标志物可重复性的主要科学家迅速做出反应,确定了透明度的需要,并提出了实现透明度的工具。尽管基因组测序仍然是一项科学壮举,但它所激发的生物学中的开放科学运动可以说是人类基因组计划对科学本身的贡献。
4. 提出没有答案的问题是有价值的
许多人认为假设检验是科学方法的决定性因素。许多人批评基于基因组学的调查,包括基因组测序,是 "钓鱼式的调查"或 "无假设的研究"。但事实是,我们通常需要一些数据或观察来开始制定一个假说。基因组学所促成的那种探索性研究使我们能够提出没有基因组规模的数据就无法设想的问题。这种方法并非没有先例,当新技术开辟了观察世界的新途径时,往往会出现这种情况。当伽利略-伽利莱制造了一架望远镜并将其转向木星时,他不可能假设木星有卫星,但观察到木星拥有四个明显围绕该行星运行的卫星,最终使他得出结论: 尼古拉斯-哥白尼的日心说模型是正确的,所有的行星,包括地球,都围绕太阳运行。同样,HGP之前也没有办法假设基因组中编码的25,000个基因是什么,或者乳腺癌有一套由表达模式定义的亚型,并与临床亚型相关。但掌握了乳腺癌的分子亚型后,人们就可以假设是什么生物过程驱动和区分不同的亚型,更重要的是,人们可以设计特定的药物来治疗它们。做科学家最大的乐趣之一就是研究没有人知道答案的问题,但这些问题往往一开始就知道,只是不知道答案,而基因组学为我们打开了一个研究问题的世界。
如果我们考察所有事实的集合,我们可以根据对它们的了解,把它们分成几个子集。有一些已知的事实,也就是我们认为我们知道和理解的事情。然后是已知的未知数,在科学中,这些未知数是最适合于假设驱动来研究问题。然后是未知的未知因素——我们甚至还不知道我们需要知道的事情。当基因组被测序后,精准医疗的概念即我们根据病人的种系或体细胞基因组来匹配治疗,成为一个优先事项,特别是在癌症等疾病方面(图4)。寻找靶点、开发和测试治疗方法,以及了解为什么这些治疗方法不是对每个携带特定目标基因突变的人都起作用,需要结合对与疾病过程相关的生物学的了解,进行探索和假设混合驱动的研究。
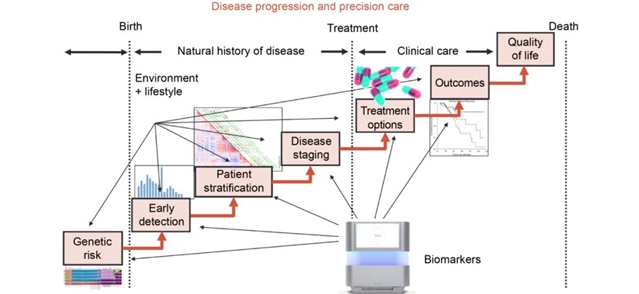
图4. 精准医疗通常被认为是HGP的终极目标
虽然做探索性研究可以是一种数据驱动的工作,但这并不意味着我们采取的方法可以忽视实验设计和分析的基本原理。我们是否有足够大的样本量来得出一个有意义的结论?我们能制定一个有意义的验证策略吗?我们的检测中是否有潜在的混杂因素,会以某种方式偏离结果?如果我们发现了什么,我们会质疑结果并寻找潜在的偏差和虚假的关联吗?做得好的发现驱动型科学并不是一种懒惰的研究方法,它实际上需要一定程度的严格分析,而这种分析在证明假设的竞赛中有时会被忽视。
基因组学的成功是被我们发现了预料之外的东西。这种发现的喜悦,发现以前没有人知道的事情是作为一个科学家的价值所在。作者认为,基因组的前二十年说明技术很可能会给人们提供数据来探索、提出假设、并验证这些假设。基因组学不是专注于狭义的问题,而是打开了未被探索的世界,让我们有机会成为未知领域的探索者。作为一名科学家,没有什么能比调查我们所知甚少的现象, 或回答长期以来我们无法回答的问题更让人感到高兴。
5. 总结
基因组学改变了过去二十年的健康和生物医学研究,无论是直接使用基因组序列本身、基因目录,还是由基因组计划催生的技术,今天生物学的几乎每一项研究都以某种方式依赖于基因组学。我们已经绘制了无数疾病的图谱,开始解开细胞中的调节过程,发现了新的疗法,对微生物组进行了采样,追踪了人类向非洲的进化和在全球的迁移,并开始沿着对相当一部分人类人口进行测序的道路。基因组学已经实现了无创产前检测,被用来监测COVID-19大流行,并正在慢慢进入常规医疗检测。测序也不限于人类--从生态学到植物科学,基因组学研究是推动该领域发展的重要工具。基因组学甚至正在进入我们更广泛的文化。当第一个基因组在2001年被测序时,人们几乎可以把基因组学想象成一种强大的调查方法,但其潜在的应用是有限的。到2009年,人们已经理解到基因组在精准治疗方面的威力,但做一个肿瘤亚型筛查可能要倾家荡产。到了今天,信用卡就能付得起基因组测序的账单,数据存储在云端,可以用任何开源工具来分析。
尽管我们在基因组科学方面取得了进展,但仍有许多工作要做。我们仍在努力了解我们的基因组是如何造就人类的,更重要的是模拟疾病风险及其发展轨迹,以及预测对治疗的反应。作者认为自己能从事这些基本问题的研究,虽然有时会感叹在有生之年我们无法回答所有的问题,但仍然十分陶醉于此。按照作家彼得-德-弗里斯的说法,"基因组就像一个有密码的保险箱, 但是密码被锁在了保险箱里",而作者仍然在饶有兴致地尝试打开这个保险箱。
QB期刊纪念HGP20周年系列文章
温馨提示:点击题目进入文章
1.美国两院院士Michael S. Waterman教授分享HGP早期历史
2.国际权威生信专家Michael Q. Zhang教授分享自己研究历程和学科发展思考
3.陈润生院士回顾我国早期生物信息学的发展
4.杨焕明院士讲述HGP发展的三个阶段、三大影响及中国对HGP的三个贡献
5.Andrew F. Neuwald教授对从基因组数据中获取生物学信息的思考
6.Jinghui Zhang教授回忆自己参加组装和分析首个人类基因组序列的故事
7.赵宏宇教授分享关于统计建模在表征人类疾病和性状遗传中的作用
8.Zhaohui S. Qin教授分享自己对HGP带来的“财富”、未来机遇及挑战的见解
Quantitative Biology期刊介绍
Quantitative Biology (QB)期刊是由清华大学、北京大学、高教出版社联合创办的全英文学术期刊。QB主要刊登生物信息学、计算生物学、系统生物学、理论生物学和合成生物学的最新研究成果和前沿进展,并为 与计算机、数学、物理等交叉研究领域打造一个学术水平高、可读性强、具有全球影响力的交叉学科期刊品牌。

《前沿》系列英文学术期刊
由教育部主管、高等教育出版社主办的《前沿》(Frontiers)系列英文学术期刊,于2006年正式创刊,以网络版和印刷版向全球发行。系列期刊包括基础科学、 、工程技术和人文社会科学四个主题,是我国覆盖学科最广泛的英文学术期刊群,其中13种被SCI收录,其他也被A&HCI、Ei、MEDLINE或相应学科国际权威检索系统收录,具有一定的国际学术影响力。系列期刊采用在线优先出版方式,保证文章以最快速度发表。
中国学术前沿期刊网
http://journal.hep.com.cn

特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。













